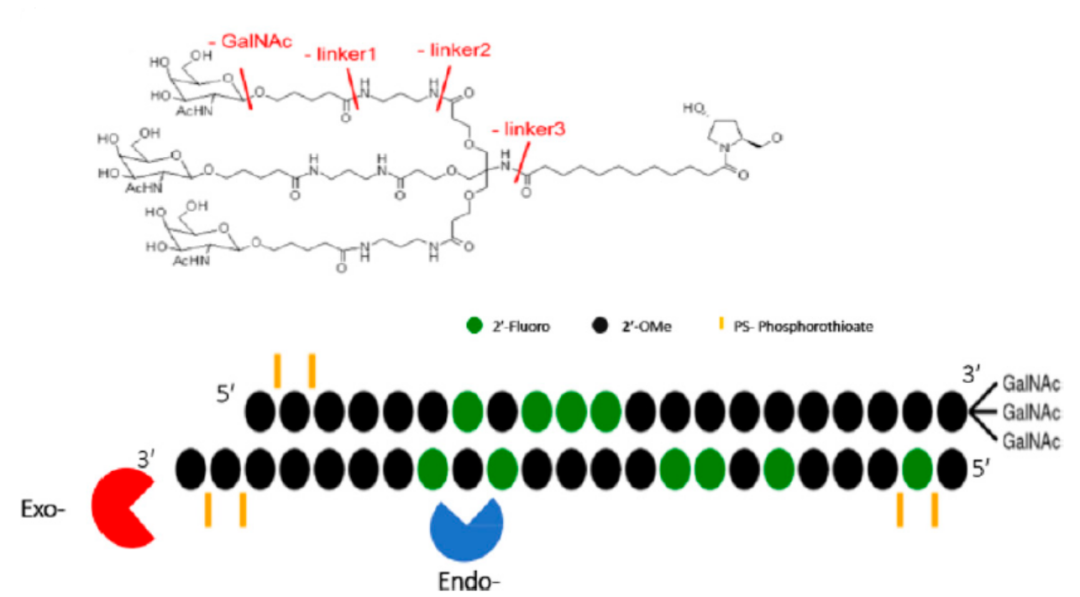
寡核苷酸藥物研發(fā)近年來發(fā)展迅速,目前FDA已批準 10款反義核苷酸(ASO)和5款siRNA藥物上市。在寡核苷酸藥物研發(fā)過(guò)程中,遞藥系統起(qǐ)着關鍵作用,其中GalNAc (N-乙酰化的半乳糖胺)直接與寡核苷酸結合或將(jiāng)其修飾到特定的遞送系統作爲靶向(xiàng)部分,以nM級高親和性結合ASGPR,在蛋白介導作用下將(jiāng)GalNAc及核酸攝取進(jìn)入肝細胞,是當前最常用的小核酸藥物遞送系統之一。
最近上市的三款siRNA藥物Givosiran(GIBLAARI)、Inclisiran(Leqvio)及Lumasiran(Oxlumo)均采用GalNAc修飾,通過(guò)將(jiāng)GalNAc以三價态的方式共價偶聯至核酸3´末端構成(chéng),增加GalNAc與ASGPR的高親和力,從而提高靶向(xiàng)性。
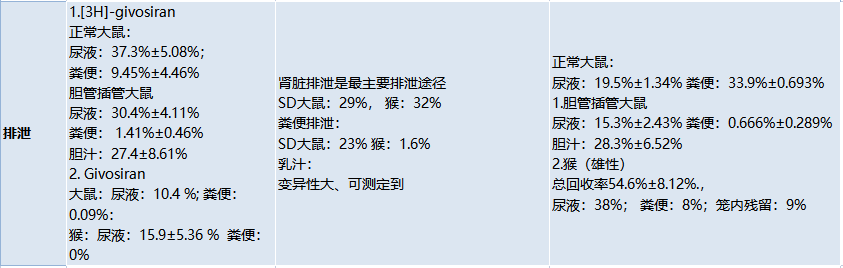
鼎泰團隊,對(duì)已上市的三款GalNAc-siRNA藥物ADME性質進(jìn)行了對(duì)比,如下表。
表1.已上市GalNAc-siRNA藥物ADME性質對(duì)比(上下滑動查看)
近期,Alnylam Pharmaceuticals研究人員在Drug Metabolism and Disposition上發(fā)表相關綜述。分享GalNAc-siRNA産品非臨床吸收、分布、代謝以及排洩(ADME)數據在不同種(zhǒng)屬間的一緻性,非臨床向(xiàng)臨床數據的轉化、臨床數據可預測性等;同時提出,在缺少臨床肝髒PK特征時,可優化臨床有效、安全劑量。
1、GalNAc-siRNAs藥物在體内循環系統中維持雙鏈結構
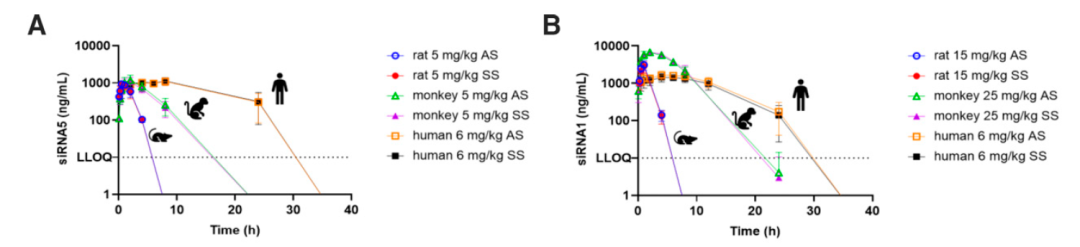
雙鏈在循環系統中穩定存在,單鏈快速地經(jīng)核酸酶代謝。體内研究顯示:在大鼠、食蟹猴及人血漿中AS鏈、SS鏈幾乎以1:1存在,如圖2所示。
GalNAc-siRNAs 在循環系統中以雙鏈形式存在,測定的單鏈AS鏈濃度水平能(néng)夠反映出完整的雙鏈濃度水平。
2、吸收(Absorption)
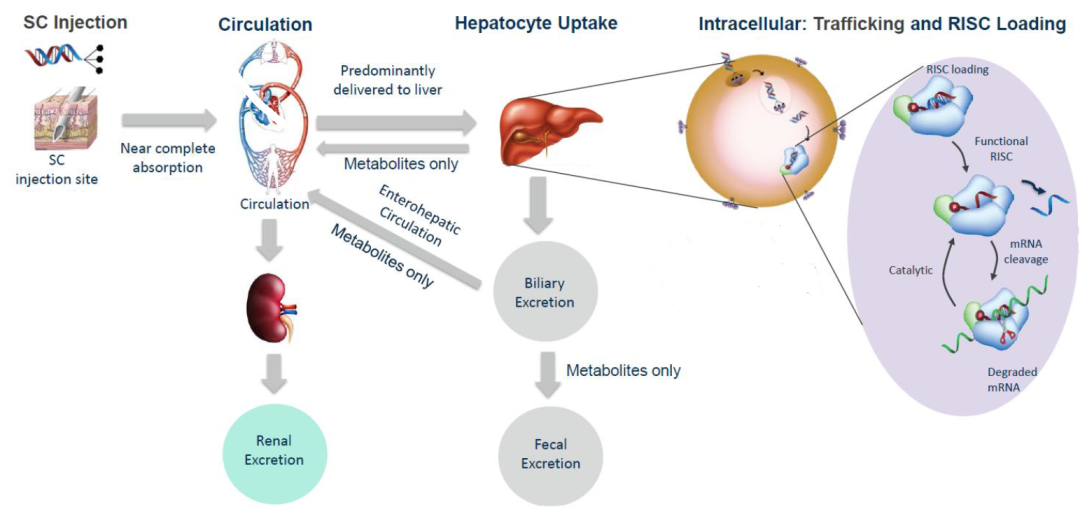
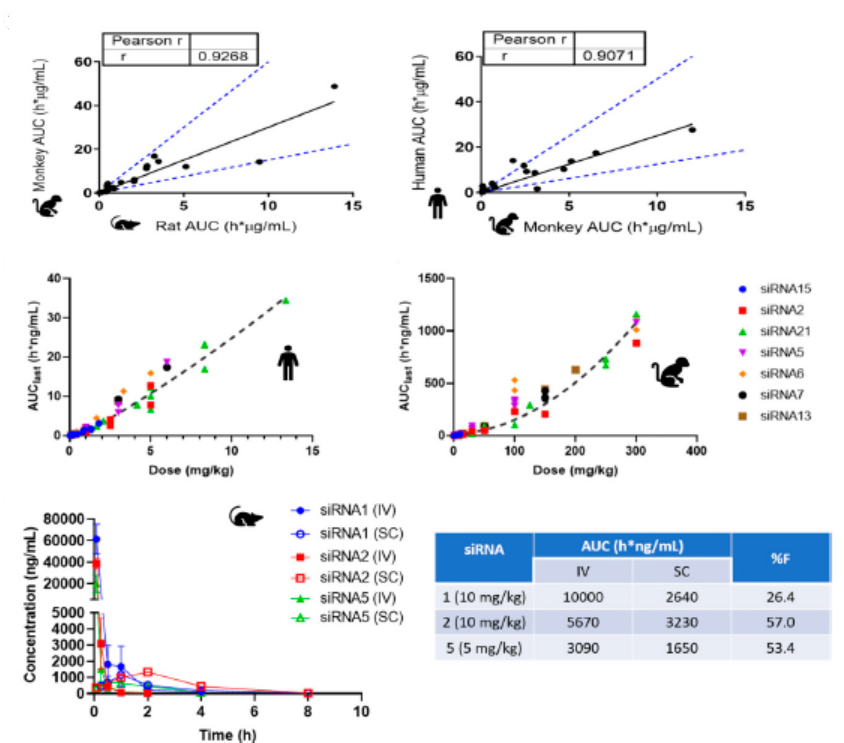
GalNAc-siRNAs藥物皮下注射給藥後(hòu),藥物會迅速吸收入血,從血漿中快速清除(主要由ASGPR介導快速被肝攝取)。給藥劑量低于10 mg/kg劑量時,在大鼠、食蟹猴以及人血漿中PK暴露量呈劑量比例增加,兩(liǎng)者相關性較好(hǎo),在人血漿中可通過(guò)異速縮放進(jìn)行預測;給藥劑量大于10mg/kg時,可能(néng)由于ASGPR介導的攝取清除的飽和的暫時飽和,血漿暴露增加,血漿中、藥物暴露量水平可能(néng)會高于劑量比例增加。如圖3所示。
GalNAc-siRNAs藥物生物利用度變異很大,在考察生物利用度時,需考慮劑量盡可能(néng)低避免ASGPR介導的飽和,同時劑量盡可能(néng)高以便于AUC能(néng)夠準确代表血漿暴露量。ASGPR介導快速分布于肝髒組織,GalNAc-siRNAs在血漿在暴露是有限的。因此,劑量設定時要相對(duì)較高的劑量水平來确保檢測足夠長(cháng)時間來表征血漿PK參數,尤其在靜脈輸注時,更需考慮最佳劑量及優化采血時間點。
3、分布(Distribution)
一般情況下,皮下給予藥效劑量後(hòu),在大鼠肝髒中暴露約63%藥物劑量,在猴肝髒中約87%藥物暴露。皮下注射(SC)給藥時,随給藥劑量增加,在肝髒分布反而減少後(hòu),但較靜脈注射(IV)給藥分布更高。
ASGPR主要在肝髒中表達。QWBA數據顯示:GalNAc-siRNAs藥物,主要分布于肝髒,肝外組織中如腎髒、淋巴結、腎上腺、胰腺、空腸和骨髓分布較少,而腦和心髒中藥物分布幾乎可忽略不計。
皮下注射時,少量藥物(<10%)滞留于注射部位或淋巴系統,從而限制延遲釋放到體循環的高濃度的可能(néng)性。
4、代謝(Metabolism)
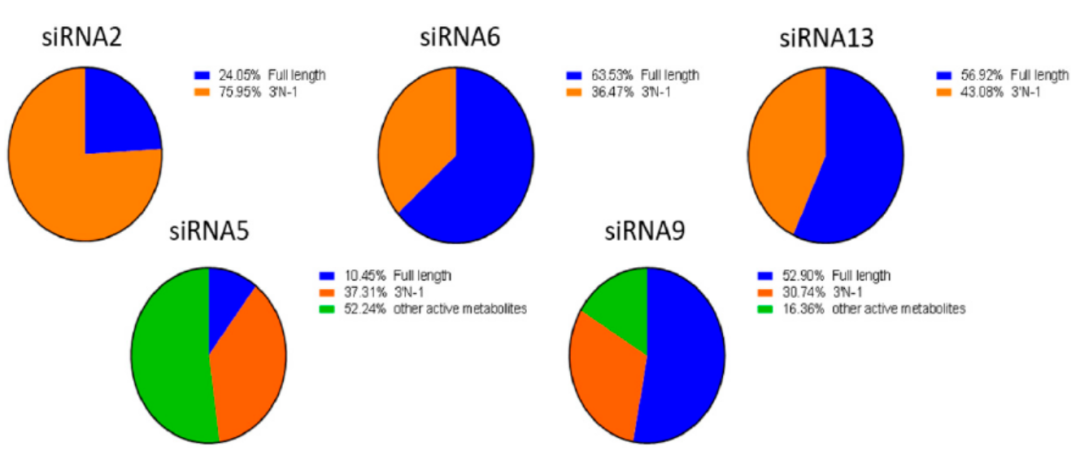
給予GalNAc-siRNAs藥物,GalNAc片段在溶酶體運輸過(guò)程中迅速被β- N -乙酰氨基葡萄糖酶清除、連接子結構進(jìn)一步被肝細胞中的酰胺酶代謝、雙鏈siRNA通過(guò)血液和組織中核酸内切酶和核酸外切酶降解代謝。核酸外切酶作用于鏈的末端,導緻單核苷酸的釋放,而内切酶作用于鏈内導緻鏈長(cháng)度改變。GalNAc-siRNAs代謝特征如圖4所示。
與完整siRNA相比,AS(N-1)3´可能(néng)與母藥活性相當,是最主要的活性代謝産物,5´核酸的丢失可能(néng)導緻其成(chéng)爲非活性代謝産物。主要代謝路徑如圖5所示。
5、排洩(Excretion)
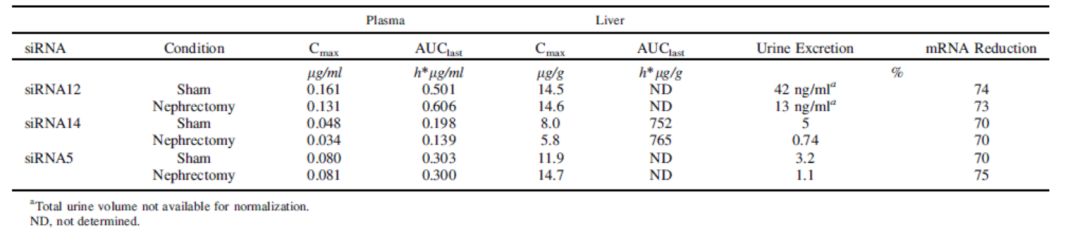
GalNAc-siRNAs藥物主要通過(guò)腎髒排洩。腎損傷時,腎髒排洩減少、血漿中暴露量可能(néng)暫時增加,但肝髒中PK(Cmax, AUC)或PD效果(mRNA Reduction )不會産生顯著影響。
6、肝靶組織PK-PD模型
GalNAc-siRNA藥物PK-PD模型建立的難點在于:一是血漿PK和藥物的PD并沒(méi)有直接的相關性。二是血漿半衰期與PD發(fā)揮作用時間的脫節。即siRNA 藥物的血漿峰濃度和最大效應之間通常存在一定的滞後(hòu),血漿動力學(xué)過(guò)程和靶組織中效應動力學(xué)過(guò)程在時間上不一緻即短時間的血漿暴露可以在組織中産生長(cháng)期的效應。
GalNAc-siRNAs藥物旨在肝靶向(xiàng)分布,PD作用效果(靶細胞中基因沉默效應)與肝半衰期及肝髒或RISC中結合siRNA的暴露量(AS鏈的數目)有關。因此,肝半衰期及RISC-siRNA數據,可特征性标準GalNAc-siRNAs藥物PK。建立基于血漿中靶蛋白的減少量(PD)與RISC-負載的siRNA濃度(PK)的PK-PD模型,通過(guò)異速縮放預測人體有效劑量。
結語
目前,GalNAc-siRNA藥物ADME和PK/PD特性已在體内、體外得到充分研究,GalNAc-siRNA在不同種(zhǒng)屬間具有一緻的ADME和PK/PD特性。在缺少直接獲得臨床肝髒PK和PD數據的情況下,非臨床研究(ADME)可爲臨床劑量和給藥方案提供依據。
參考資料:
[1] Mcdougall R , Ramsden D , Agarwal S , et al. The Nonclinical Disposition and Pharmacokinetic/ Pharmacodynamic Properties of N-Acetylgalactosamine-Conjugated Small Interfering RNA Are Highly Predictable and Build Confidence in Translation to Human[J]. Drug Metabolism and Disposition, 2022(6):50.
[2] Assessment report: Givlaari International non-proprietary name: givosiran Committee for Medicinal Products for Human Use (CHMP), EMA/CHMP/70703/2020,https://www.ema.europa.eu/en/medicines/human/EPAR/givlaari
[3] Assessment repor: Oxlumo International non-proprietary name: lumasiran Committee for Medicinal Products for Human Use (CHMP), EMA/568312/2020, https://www.ema.europa.eu/en/medicines/human/EPAR/oxlumo
[4] Assessment repor: Oxlumo International non-proprietary name: lumasiran Committee for Medicinal Products for Human Use (CHMP), EMA/568312/2020, https://www.ema.europa.eu/en/medicines/human/EPAR/oxlumo
[5] Assessment report: Oxlumo International non-proprietary name: lumasiran Committee for Medicinal Products for Human Use (CHMP), EMA/568312/2020, https://www.ema.europa.eu/en/medicines/human/EPAR/oxlumo
[6] Assessment report: Leqvio International non-proprietary name: inclisiran Committee for Medicinal Products for Human Use (CHMP), EMA/696912/2020 https://www.ema.europa.eu/en/medicines/human/EPAR/leqvio






 官方微信
官方微信
 官方視頻号
官方視頻号

 法規政策
法規政策 項目管理
項目管理




 Global
Global