


上一期,鼎泰集團轉發(fā)了已上市的ADC藥物的非臨床研究的彙總整理(全球已上市ADC藥物非臨床研究一覽-1)。爲了更深入、系統地分析不同創新程度、不同适應症和作用機制的ADC藥物的非臨床開(kāi)發(fā)路徑,鼎泰團隊系統整理并歸納總結了已上市ADC藥物所開(kāi)展的支持IND和BLA的非臨床研究。 本系列分享按小分子毒素 (Payload) 將(jiāng)已上市ADC藥物分爲3類: 微管蛋白抑制劑,如MMAE、DM1、DM4 拓撲異構酶抑制劑,如DXd、SN-38 其他,如Calicheamicin 在研ADC産品payload的多樣性及占比[1] 本期分享聚焦以微管抑制劑爲Payload的已上市ADC藥物(Kadcyla®、Elahere®、Adcetris®、Polivy®、Padcev®、Tivdak®等)的非臨床研究曆程(以MMAF爲Payload 的 Blenrep®因确證性臨床試驗失敗,未進(jìn)行分析),同時結合CDE《抗體偶聯藥物非臨床研究技術指導原則》(2023年)、ICH S9、ICH R6(R1) 和 ICH M3(R2) 對(duì)相關研究内容進(jìn)行了分析。期望通過(guò)本次分享爲業内同行ADC藥物非臨床研究提供精細參考,也期待與廣大業内同行一起(qǐ)交流、探讨和提升。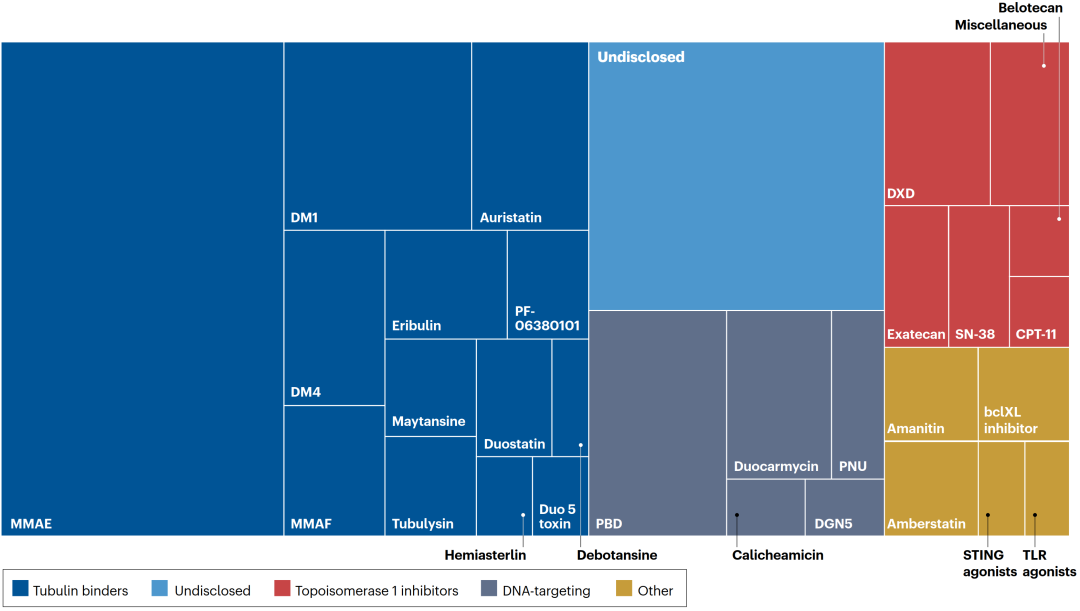
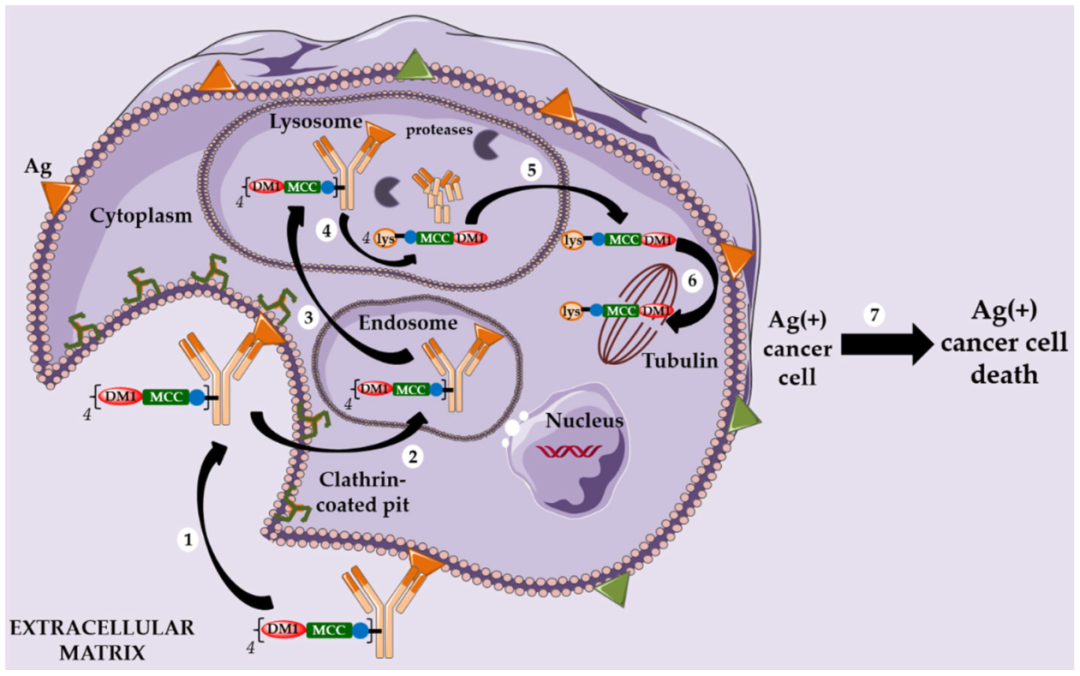
以微管蛋白抑制劑DM1爲Payload的ADC藥物的作用機制[2] 以微管蛋白抑制劑MMAE爲Payload的ADC藥物的作用機制[2]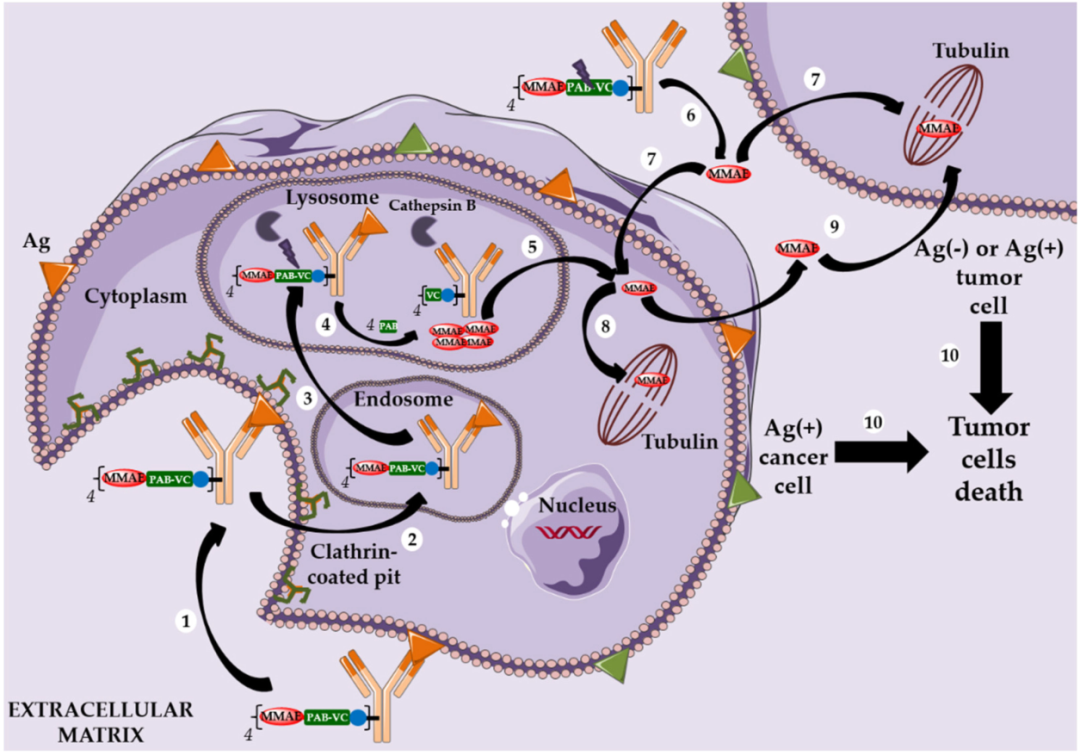
★ 目錄 ★ 01 Kadcyla®(Trastuzumab emtansine)非臨床研究内容 02 Elahere®(Mirvetuximab Soravtansine)非臨床研究内容 03 Adcetris®(Brentuximab vedotin)非臨床研究曆程 04 Polivy®(Polatuzumab vedotin-piiq)非臨床研究内容 05 Padcev®(Enfortumab vedotin-ejfv)非臨床研究内容 06 Tivdak®(Tisotumab vedotin-tftv)非臨床研究内容
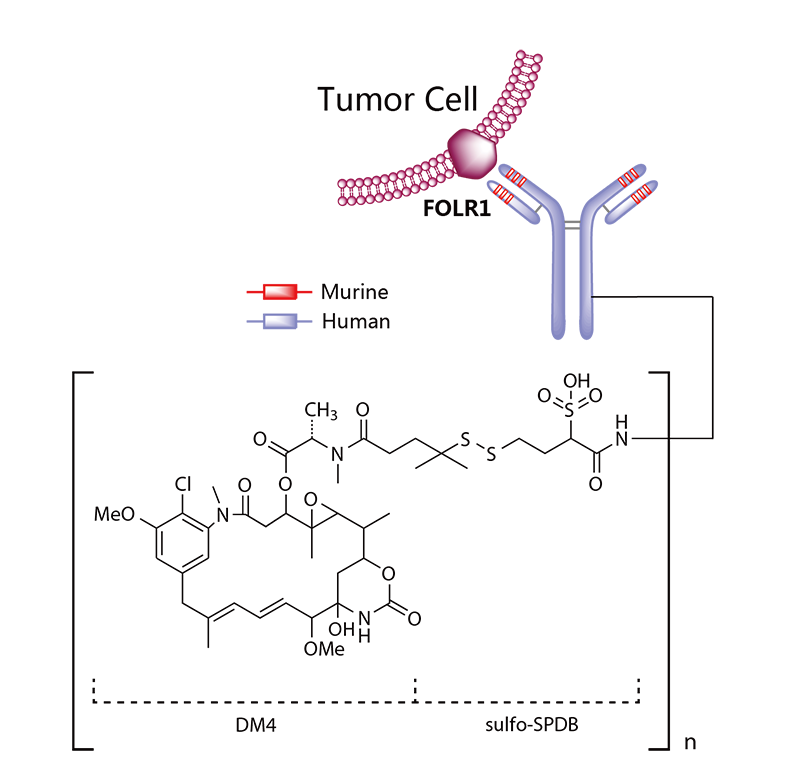
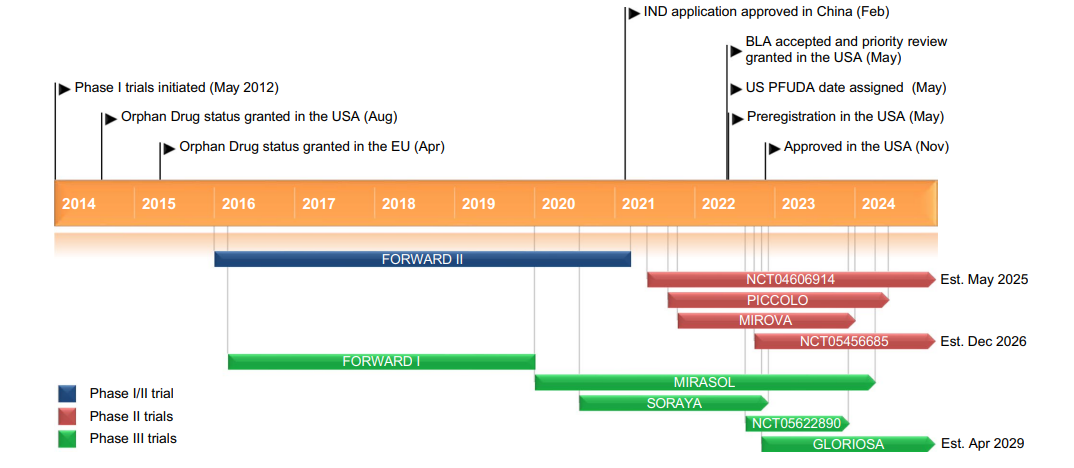
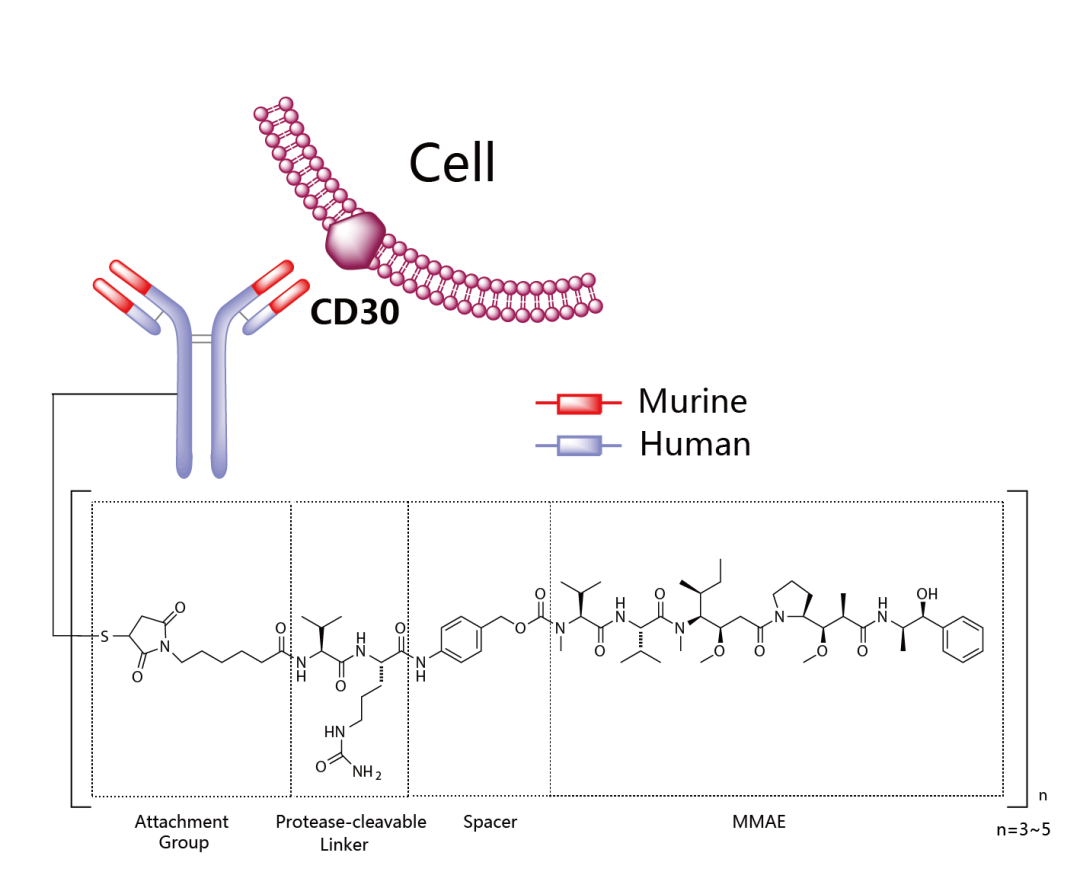
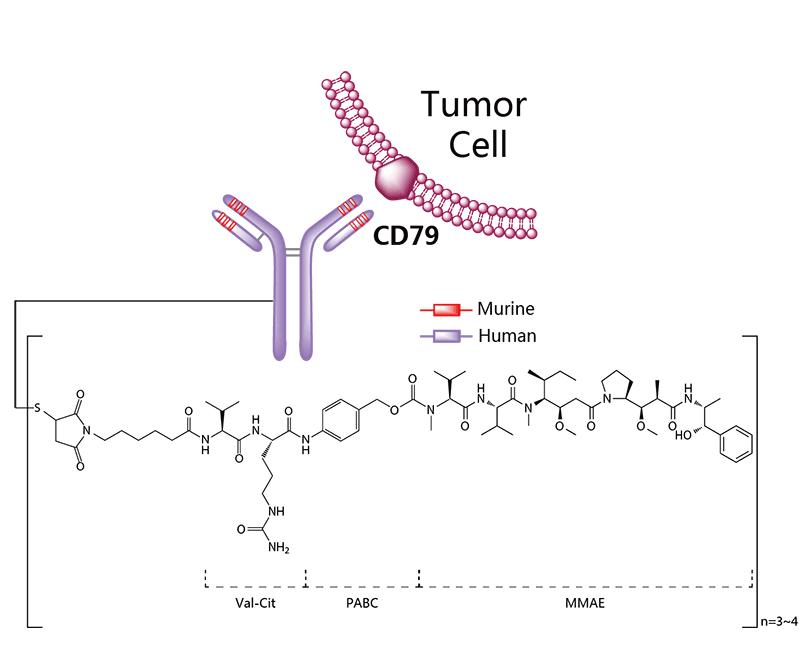
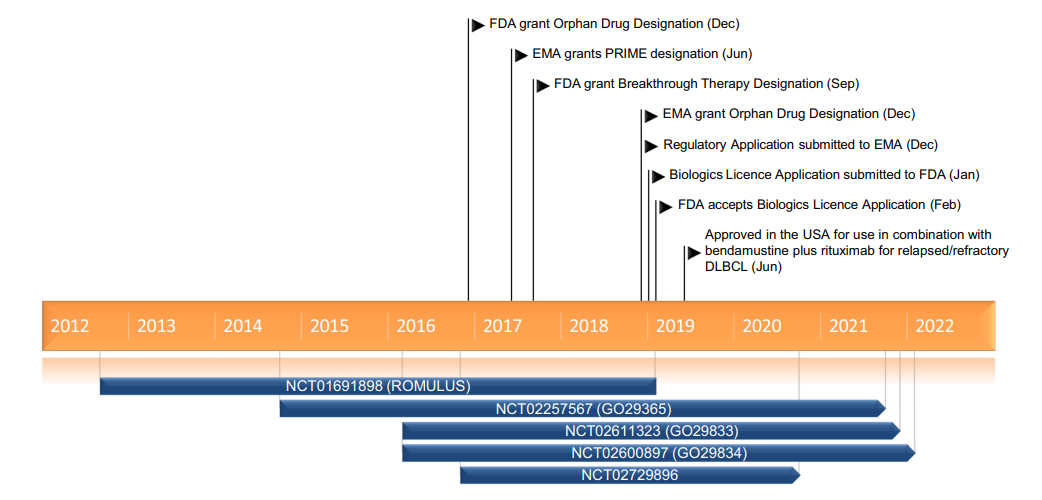
Kadcyla®(Trastuzumab emtansine) 非臨床研究内容 通過(guò)不可裂解SMCC連接子將(jiāng)靶向(xiàng)HER2的抗體(IgG1)和微管抑制劑美登素衍生物DM1連接而成(chéng)的ADC。 DAR約爲 3.5,分子量約 148.781 kDa。 已獲批适應症:用于治療先前接受曲妥珠單抗和紫杉烷單獨或聯合治療的HER2陽性轉移性乳腺癌患者。 用法用量:臨床推薦劑量爲 3.6 mg/kg,靜脈輸注,每3周1次。 Kadcyla®(Trastuzumab emtansine)化學(xué)結構,圖源藥渡 Kadcyla®(Trastuzumab emtansine,T-DM1)開(kāi)發(fā)的關鍵裡(lǐ)程碑,圖源藥渡 2005年12月16日,向(xiàng)FDA提交IND申請。 2010年7月6日,提交BLA申請,被拒絕批準後(hòu)于2012年再次提交BLA申請;2013年02月22日,由FDA首次批準上市。 | 支持首次人體試驗(FIH)IND 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 | 支持後(hòu)續 IND 和 BLA 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 由于DM1具有遺傳毒性和對(duì)快速分裂細胞的細胞毒性,且Trastuzumab也存在生殖毒性,因此T-DM1的生殖毒性更大。回顧研究顯示,6名接受Trastuzumab治療的孕婦中有3名出現羊水缺乏症狀[3]。結合 ICH S9 和 Trastuzumab 的生殖毒性試驗結果,FDA認爲上述研究足以支持T-DM1的上市批準。 Elahere®(Mirvetuximab Soravtansine)非臨床研究内容 由抗FRα單克隆抗體(IgG1)通過(guò)可切割連接子與微管抑制劑美登素衍生物DM4連接組成(chéng)。 DAR約爲3.4,抗體分子量約150 kDa。 已獲批的适應症:FRα陽性、鉑耐藥上皮性卵巢癌、輸卵管癌或原發(fā)性腹膜癌。 用法用量:推薦劑量爲 6mg /kg,根據調整後(hòu)的理想體重(AIBW)計算,每3周靜脈輸注一次,直至疾病進(jìn)展或出現不可接受的毒性。僅在5%葡萄糖注射液(USP)中稀釋後(hòu)靜脈輸注。 Elahere®(Mirvetuximab Soravtansine)化學(xué)結構,圖源藥渡 Elahere®(Mirvetuximab Soravtansine)開(kāi)發(fā)的關鍵裡(lǐ)程碑[4] 2012年5月啓動1期臨床試驗。 2022年3月在美國(guó)提交BLA申請,2022年11月14日,獲得加速批準上市。該上市申請基于如下關鍵性臨床試驗: 在一項單臂試驗臨床試驗 0417(NCT04296890) 中,對(duì)106例先前接受過(guò) ≤3 種(zhǒng)治療方案的FRα陽性、鉑耐藥複發(fā)上皮性卵巢癌、輸卵管癌或原發(fā)性腹膜癌患者進(jìn)行了療效評估,所有患者均需接受過(guò)Bevacizumab治療。主要終點爲按照 RECIST v1.1 評價的ORR 和 DCR。結果顯示,ORR爲 31.7%(22.9,41.6),中位DCR爲6.9個月。 | 支持首次人體試驗(FIH)IND 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 | 支持後(hòu)續 IND 和 BLA 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 根據ICH S9指南,晚期癌症适應症不需要開(kāi)展緻癌性研究。Elahere®的細胞毒性成(chéng)分 DM4 破壞微管功能(néng),具有遺傳毒性,對(duì)活躍分裂的細胞具有毒性,這(zhè)表明它有可能(néng)導緻胚胎毒性和緻畸性,未對(duì)Mirvetuximab soravtansine進(jìn)行生殖和發(fā)育毒性試驗。 Adcetris®(Brentuximab vedotin) 非臨床研究曆程 通過(guò)蛋白酶可切割連接子將(jiāng)靶向(xiàng)CD30的單克隆抗體(IgG1)和微管蛋白抑制劑單甲基澳瑞他汀E(MMAE)連接而成(chéng)的ADC。 DAR約爲 4,分子量約153 kDa。 已獲批的适應症:自體幹細胞移植後(hòu)複發(fā)的霍奇金淋巴瘤(HL),以及複發(fā)或難治性全身間變性大細胞淋巴瘤(sALCL)。 用法用量:臨床推薦劑量爲1.8 mg/kg,靜脈輸注30分鍾以上,每3周1次。 Adcetris®(Brentuximab vedotin)化學(xué)結構,圖源藥渡 Adcetris®(Brentuximab vedotin,SGN-35)開(kāi)發(fā)的關鍵裡(lǐ)程碑,圖源藥渡 2006年,啓動首次人體試驗(FIH),具體獲得IND批準的時間未知。 2011年2月28日,向(xiàng)FDA提交BLA申請;2011年5月2日,獲得優先審評;8月29日獲批上市。該上市申請基于2項關鍵性臨床試驗: 在一項開(kāi)放、單臂、多中心臨床試驗(NCT00848926)中,評價了Adcetris®對(duì)自身幹細胞移植後(hòu)複發(fā)HL患者的療效。該研究共納入102例患者,主要終點是客觀緩解率(ORR)和緩解持續時間(DOR)。結果顯示,接受治療的患者ORR爲 73%,中位DOR爲 6.7 個月。 在另一項開(kāi)放、單臂、多中心臨床II期試驗(NCT00866047)中,評價了Adcetris®對(duì)複發(fā)性sALCL患者的療效。該研究共納入58例患者,主要終點是ORR 和 DOR。研究顯示,接受治療的患者ORR爲 86%,中位DOR爲 12.6個月。 | 支持首次人體試驗(FIH)IND 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 | 支持後(hòu)續 IND 和 BLA 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 Polivy®(Polatuzumab vedotin-piiq) 非臨床研究内容 通過(guò)蛋白可切割連接子(MC-vcPAB)將(jiāng)靶向(xiàng)CD79b的抗體(IgG1)和微管蛋白抑制劑MMAE連接而成(chéng)的ADC。 DAR約爲 3.5,分子量約150 kDa。 已獲批的适應症:複發(fā)或難治性彌漫性大B細胞淋巴瘤(DLBCL)。 用法用量:臨床推薦劑量爲1.8 mg/kg,靜脈輸注90分鍾以上,每21天一次,共6個療程。 Polivy® (Polatuzumab vedotin-piiq)化學(xué)結構,圖源藥渡 Polivy®(Polatuzumab vedotin-piiq)開(kāi)發(fā)的關鍵裡(lǐ)程碑[5] 根據文獻分析,推測在2010年前後(hòu)提交IND申請。于2014年曾因嚴重的毒性反應而被叫(jiào)停試驗(partial clinical hold)。 2018年12月19日,向(xiàng)FDA提交BLA申請,并于2019年6月10日FDA加速批準上市,其基于1項關鍵的臨床試驗: 在一項開(kāi)放标簽、多中心臨床試驗 GO29365(NCT02257567),納入了 80 名至少經(jīng)過(guò)一線治療的複發(fā)或難治性DLBCL患者,随機(1:1)接受Polatuzumab vedotin-piiq聯合Bendamustine 和 Rituximab(P+BR) 或 BR,21天爲一個給藥周期,共給藥6周期。Polatuzumab vedotin-piiq,在第1周期的第2天和随後(hòu)周期的第1天靜脈注射(1.8 mg/kg);Bendamustine在第1周期的第2天和第3天以及随後(hòu)周期的第1天和第2天靜脈注射(90mg/m2);Rituximab在每個周期的第1天靜脈注射(375 mg/m2)。治療結束時,P+BR和BR組的CR分别爲 40% (95% CI: 25-57%) vs 18%(95% CI: 7-33%),ORR分别爲 63% vs 25%。在25例對(duì)P+BR達到CR或PR的患者中,16例(64%)的DCR至少爲6個月,12例(48%)的DCR至少爲12個月。該适應症根據主要終點CR的結果獲得加速批準。 | 支持首次人體試驗(FIH)IND 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 | 支持後(hòu)續 IND 和 BLA 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰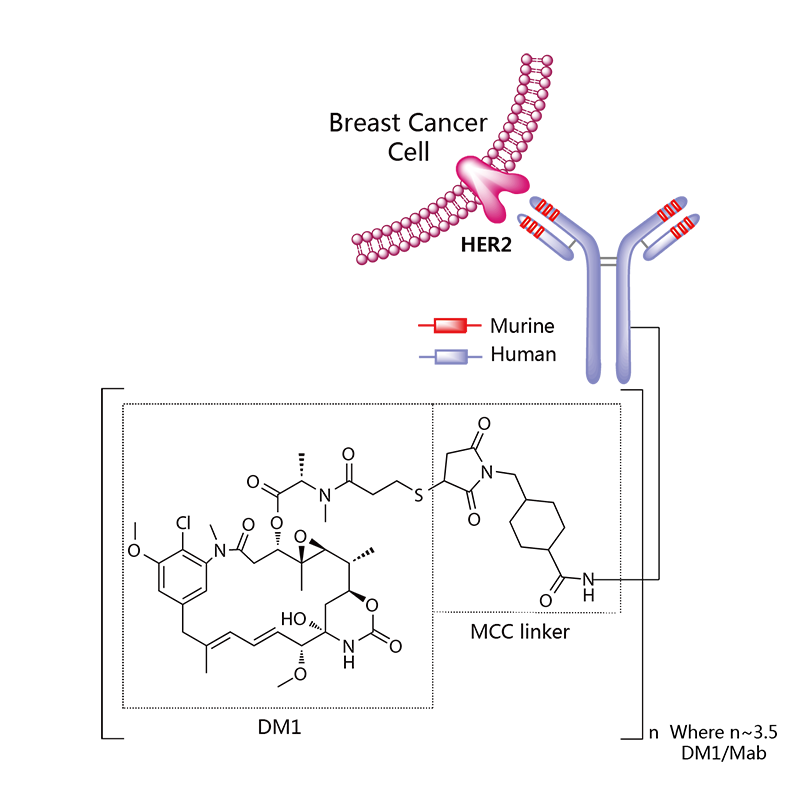









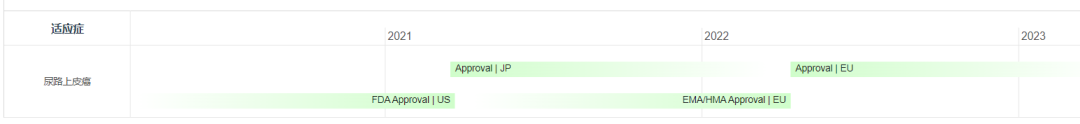
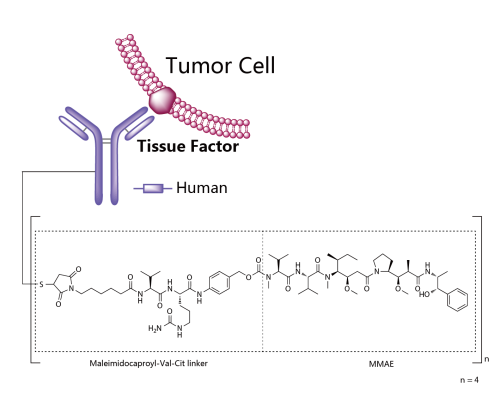
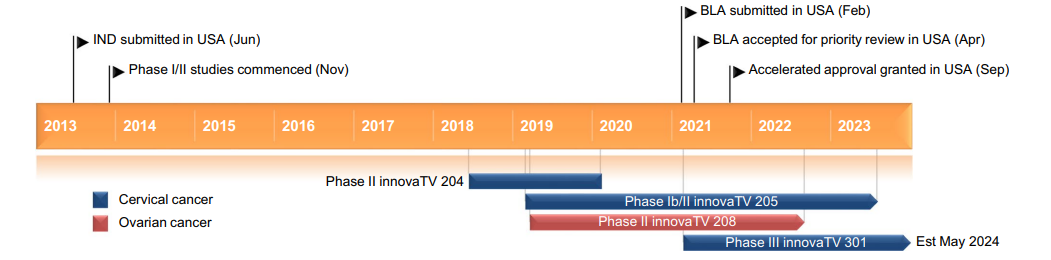
Padcev®(Enfortumab vedotin-ejfv) 非臨床研究内容 通過(guò)馬酰亞胺丙基缬氨酸-瓜氨酸(protease-cleavable maleimidocaproyl valine-citrulline, vc)連接子將(jiāng)靶向(xiàng)Nectin -4的抗體(Ig G1)和微管抑制劑MMAE連接的ADC。 DAR約爲 3.8 ,分子量約152 kDa。 已獲批的适應症:局部晚期或轉移性尿路上皮癌,患者先前接受過(guò)程序性死亡受體-1(PD-1)或程序性死亡配體1(PD-L1)抑制劑,以及新輔助/輔助、局部晚期或轉移性環境下的含鉑化療。 用法用量:1.25 mg/kg(最大劑量125mg),在28天周期的第1、8 和 15天靜脈輸注30分鍾,直至疾病進(jìn)展或不可接受的毒性。 Padcev®(Enfortumab vedotin-ejfv)結構,圖源藥渡 Padcev®(Enfortumab vedotin-ejfv)開(kāi)發(fā)的關鍵裡(lǐ)程碑,圖源藥渡 2012年12月28日,在FDA首次遞交IND申請(IND116360)。在此之前,已啓動代号爲AGS-22M6E-11-1的首次人體試驗(FIH)。 2019年7月15日提交BLA申請,2019年12月18日獲得FDA加速批準上市。Padcev®(Enfortumab vedotin-ejfv)的獲批上市基于如下關鍵性臨床試驗: 在一項單臂、多中心試驗 EV-201(NCT03219333) 中,納入 125 例既往接受過(guò) PD-1 或 PD-L1 單抗和含鉑化療的局部晚期或轉移性尿路上皮癌患者。在爲期28天的治療周期中的第1、8和15天給予 1.25 mg/kg Enfortumab vedotin-ejfv,直到疾病進(jìn)展或不可耐受的毒性。主要終點爲ORR和DCR。ORR爲44% (95% CI: 35.1, 53.2),其中CR爲12%,PR爲32%;DCR爲7.6個月(95% CI: 6.3)。該适應症的完全批準取決于确證性臨床試驗的結果。 | 支持首次人體試驗(FIH)IND 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 | 支持後(hòu)續 IND 和 BLA 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 小分子毒素MMAE的ADME部分試驗參考 BLA 125388(Brentuximab vedotin),未單獨開(kāi)展,内容包括:MMAE的血漿蛋白結合試驗,[3H]-MMAE的CYP酶代謝表型、[3H]- MMAE在大鼠、猴子和人肝細胞中的代謝産物鑒定和MMAE對(duì)人細胞色素P450誘導和抑制試驗。MMAE的光安全性試驗參考BLA 761121(Polatuzumab vedotin-piiq) 。 Tivdak®(Tisotumab vedotin-tftv) 非臨床研究内容 由TF特異性人IgG1- κ單克隆抗體通過(guò)蛋白酶可切割缬氨酸-瓜氨酸(vc)連接子與微管抑制劑MMAE結合。 DAR約爲 4,分子量約153 kDa。 已獲批的适應症:複發(fā)性或轉移性宮頸癌。 用法用量:TIVDAK的推薦劑量爲2mg /kg(最多200mg),每3周靜脈輸注 30min,直至疾病進(jìn)展或出現不可接受的毒性。 Tivdak® (Tisotumab vedotin-tftv)化學(xué)結構,圖源藥渡 Tivdak® (tisotumab vedotin-tftv)開(kāi)發(fā)的關鍵裡(lǐ)程碑[6] 2013年7月(GEN701)首次提交IND申請。 2021年03提交BLA,并于2021年09月20日 FDA加速批準上市。該上市申請基于如下關鍵性臨床試驗: 在一項開(kāi)放标簽、多中心、單臂臨床試驗(InnovaTV 204,NCT03438396)中,共納入101例接受過(guò)不超過(guò)2種(zhǒng)系統治療的複發(fā)或轉移性宮頸癌患者,且至少一種(zhǒng)含鉑化療,69%的患者接受過(guò)Bevacizumab全身治療。患者每3周接受2mg /kg的Tisotumab vedotin-tftv治療,直至疾病進(jìn)展或出現不可接受的毒性。ORR爲24% (95% CI: 15.9%, 33.3%),中位DOR爲8.3個月(95% CI: 4.2)。 | 支持首次人體試驗(FIH)IND 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 | 支持後(hòu)續 IND 和 BLA 申請的非臨床研究 鼎泰團隊自制圖,點擊查看更清晰 小分子毒素MMAE的ADME部分試驗參考BLA 125388(Brentuximab vedotin),未單獨開(kāi)展,内容包括:MMAE的血漿蛋白結合試驗,[3H]-MMAE的CYP酶代謝表型、[3H]- MMAE在大鼠、猴子和人肝細胞中的代謝産物鑒定、 cAC10-vc-3H-MMAE或[3H]-MMAE在大鼠體内的排洩、物質平衡和藥代動力學(xué)研究。 未針對(duì) Tisotumab vedotin-tftv 開(kāi)展胚胎-胎仔發(fā)育毒試驗;MMAE(SGD-1010)胚胎-胎仔發(fā)育毒性試驗參考 BLA 125388(Brentuximab vedotin)。根據ICH S9,拟用于晚期癌症患者的,可不開(kāi)展生育能(néng)力和早期胚胎發(fā)育以及圍産期發(fā)育毒性試驗。 FDA根據申請人提交的 Brentuximab vedotin 申報資料(BLA 125388)對(duì) Tisotumab vedotin-tftv 的遺傳毒性進(jìn)行了審評,包括:MMAE細菌回複突變試驗(陰性)、MMAE小鼠淋巴瘤正向(xiàng)突變試驗(陰性)、MMAE大鼠骨髓微核試驗(陽性)。 結語 ADC藥物的非臨床研究需要考慮其複雜的組成(chéng)結構和體内行爲。抗體、連接子和小分子毒素三者之間的交互作用會影響藥物的安全性和有效性。因此,非臨床評價需要全面(miàn)而審慎。 根據ICH M3(R2),開(kāi)始支持FIH的IND申請前,進(jìn)行ADC藥物的血漿穩定性和毒素分子的血漿蛋白結合率試驗有助于了解體内行爲;針對(duì)性的體外和體内藥效學(xué)試驗有助于闡明ADC藥物的初步作用機制、潛在的臨床有效性,有時可爲适應症的選擇提供參考;對(duì)ADC藥物進(jìn)行短周期的重複給藥毒性試驗有助于獲得支持臨床試驗設計的安全性數據。如果毒素爲新分子,需要進(jìn)行單獨的毒性評價;如果該毒素分子有可參考的合法引用或公開(kāi)的非臨床數據,可借用文獻資料(如MMAE)。在臨床試驗期間,可開(kāi)展支持後(hòu)續IND申請和BLA申請的更長(cháng)周期的毒理試驗,以及爲解答前期非臨床和臨床試驗中出現的毒性而開(kāi)展的追加試驗(如眼毒性);參考ICH S9,開(kāi)展必要的生殖和發(fā)育毒性試驗,對(duì)于拟用于晚期腫瘤患者的ADC藥物,通常無需開(kāi)展緻癌性試驗。在臨床試驗期間,可根據工藝變更的等級開(kāi)展必要的非臨床研究。 由于部分試驗的起(qǐ)始時間在FDA審評報告中無法準确獲知,鼎泰團隊基于共識和經(jīng)驗進(jìn)行了分析。如有不準确之處,歡迎同行提出寶貴意見,以提高該調研報告的嚴謹性。 鑒于篇幅所限,本文重點對(duì)多個已上市ADC藥物的非臨床研究路徑進(jìn)行了彙總和複盤,尚未深入分析每個産品的非臨床策略和非臨床研究的臨床預測價值。後(hòu)續鼎泰團隊將(jiāng)選擇代表性案例,從更爲宏觀的視角進(jìn)行深度解讀。這(zhè)將(jiāng)有助于我們進(jìn)一步理解和優化ADC藥物的非臨床研究策略,爲後(hòu)續同類藥物的非臨床研究提供借鑒,從而更好(hǎo)地支持臨床試驗設計和監管審評。 下一期,我們將(jiāng)分享以拓撲異構酶抑制劑爲Payload的ADC藥物的非臨床研究内容,敬請關注。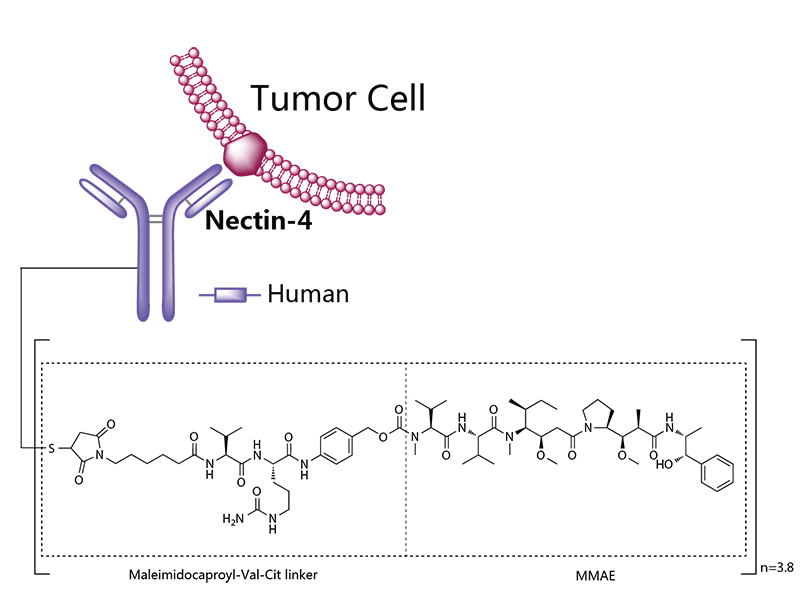




參考資料: 供稿:鼎泰集團轉化科學(xué)和藥政策略部






 官方微信
官方微信
 官方視頻号
官方視頻号

 法規政策
法規政策 項目管理
項目管理




 Global
Global
